Мышечная дистрофия Дюшенна
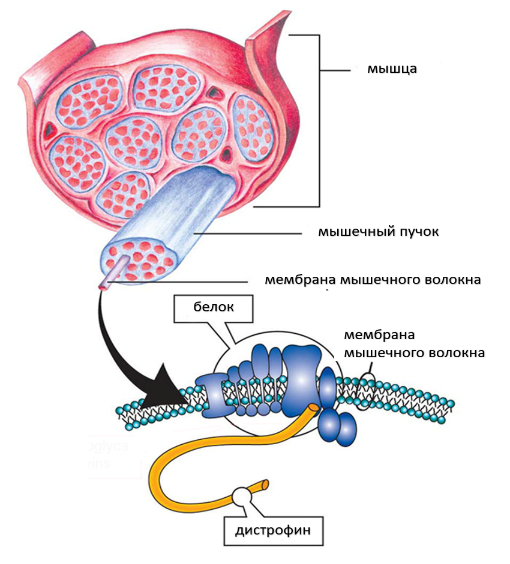
Мышечная дистрофия Дюшенна (МДД) — это генетическое заболевание, характеризующееся прогрессирующей дегенерацией мышц и их слабостью, в результате генетического дефекта белка, называемого дистрофином, который помогает сохранять мышечные клетки неповрежденными.
МДД — одно из четырех состояний, известных как дистрофинопатии. Симптомы МДД проявляются в раннем детстве, обычно в возрасте от 2 до 3 лет, до этого возраста двигательное развитие ребенка нормальное. Заболевание в первую очередь поражает мальчиков, но в редких случаях может поражать и девочек.
Распространенность МДД составляет примерно 6 на 100 000 человек.
Наследование при МДД
МДД наследуется по Х-сцепленному типу, потому что ген, который может нести вызывающую МДД мутацию, находится на Х-хромосоме. Каждый мальчик наследует Х-хромосому от матери и Y-хромосому от отца. Девочки получают две Х-хромосомы, по одной от каждого родителя.
Каждый сын, рожденный от женщины с мутацией дистрофина в одной из ее двух Х-хромосом, имеет 50-процентный шанс унаследовать дефектный ген и заболеть МДД. Каждая из ее дочерей имеет 50-процентный шанс унаследовать мутацию и стать носителем. Носители могут не иметь никаких симптомов болезни, но могут иметь ребенка с мутацией или заболеванием.
Хотя МДД часто встречается в семье, в анамнезе которой не было МДД, т.е. может внезапно родиться сын с этим заболеванием. Есть два возможных объяснения. Во-первых, генетическая мутация, приводящая к МДД, могла существовать у женщин в семье в течение нескольких поколений, о чем никто не знал. Возможно, ни один ребенок мужского пола не родился с этой болезнью, или, даже если мальчик в более раннем поколении был поражен, родственники могли не знать, какое у него заболевание.
Вторая возможность заключается в том, что у ребенка с МДД есть новая генетическая мутация, возникшая в одной из яйцеклеток его матери. Поскольку этой мутации нет в клетках крови матери, ее невозможно обнаружить стандартным тестированием на носительство.
Мужчина с МДД не может передать дефектный ген своим сыновьям, потому что он дает сыну Y-хромосому, а не X. Но он обязательно передаст ее своим дочерям, потому что каждая дочь наследует единственную X-хромосому своего отца. Тогда они станут носителями, и у каждого из их сыновей будет 50-процентная вероятность развития болезни и так далее.
Причина МДД
МДД был впервые описан французским неврологом Гийомом Бенджамином Амандом Дюшенном в 1860-х годах. В 1986 году был выявлен ген в Х-хромосоме, который в случае дефекта (мутации) приводит к МДД. В 1987 году белок, связанный с этим геном, был идентифицирован и назван дистрофином. Ген дистрофина является самым геном, идентифицированным у людей, и расположен в коротком плече Х-хромосомы в локусе Xp21.2.
Мышечная дистрофия Дюшенна возникает из-за того, что мутировавший ген DMD не может производить практически какой-либо функциональный дистрофин. Люди с генетическими мутациями мышечной дистрофией Беккера производят частично функциональный дистрофин. Недостаток белка дистрофина в мышечных клетках делает их хрупкими и легко повреждаются.
МДД имеет рецессивный характер наследования, сцепленный с Х-хромосомой, и передается от матери, которую называют носителем. Носители МДД — женщины с нормальным геном дистрофина на одной Х-хромосоме и аномальным геном дистрофина на другой Х-хромосоме. Большинство носителей МДД не имеют признаков и симптомов болезни, но некоторые могут иметь признаки заболевания. Симптомы могут варьироваться от легкой слабости скелетных мышц или поражения сердца к тяжелой слабости или сердечных нарушений и могут начаться в детстве или во взрослом возрасте.
До относительно недавнего времени мальчики с МДД обычно не доживали до подросткового возраста. Благодаря достижениям в области кардиологической и респираторной помощи, продолжительность жизни увеличивается, и многие молодые люди с МДД посещают школы, работают, женятся и заводят детей. Доживших до 30-ти лет становится все больше, чем раньше.
Симптомы МДД
Мышечная слабость — главный симптом МДД. Заболевание может начаться уже в возрасте 2 или 3 лет, сначала поражая проксимальные мышцы, а затем поражаются дистальные мышцы конечностей. У ребенка могут быть проблемы с прыжками, бегом и ходьбой. Другие симптомы включают увеличение икроножных мышц, походка вразвалочку (утиная походка) и усиление поясничного лордоза (искривление позвоночника внутрь). В дальнейшем поражаются также сердечный и дыхательные мышцы. Прогрессирующая слабость и сколиоз приводят к нарушению функции легких, в конечном итоге может вызвать острую дыхательную недостаточность.
У мальчиков с МДД часто увеличиваются икроножные мышцы — псевдогипертрофия, могут увеличиваться так же и мышцы бедер.
Слабость, связанная с мышечной дистрофией Дюшенна, избирательно поражает мышцы конечностей, расположенные близко к туловищу (проксимальные мышцы), также ноги поражаются раньше рук. Скорость роста ребенка при МДД в первые годы жизни обычно медленнее, что приводит к низкорослости.
Мальчики с МДД часто поздно начинают ходить. К школьному возрасту может показаться, что ребенок неуклюжий и часто падает. Родители также могут заметить, что детям сложно подниматься по лестнице, вставать с пола или бегать. Поднимаясь с пола, мальчики могут использовать опору для рук, чтобы подняться в вертикальное положение.
К школьному возрасту дети могут ходить на цыпочках, слегка хромая походкой, и часто падают. Чтобы сохранить равновесие, они могут выпячивать живот и расправлять плечи. Дети также испытывают трудности с поднятием рук.
Многие дети с МДД начинают пользоваться инвалидной коляской примерно до 12 лет. Переход на инвалидную коляску обычно происходит постепенно; на первых порах стул может понадобиться только для преодоления больших расстояний. Дети часто вновь обретают самостоятельность, когда полностью переходят на инвалидную коляску с электроприводом.
Пациенты с МДД часто умирают в подростковом возрасте или в возрасте 20 лет от дыхательной недостаточности или кардиомиопатии; только некоторые пациенты с МДД выживают после третьего десятилетия.
Женщины и МДД
Болезни, унаследованные по X-сцепленному рецессивному типу, в основном поражают мужчин, потому что вторая Х-хромосома обычно защищает женщин от проявлений.
Когда девочка наследует дефектный ген дистрофина от одного из родителей, она обычно также получает здоровый ген дистрофина от другого родителя, давая ей достаточно белка, чтобы защитить ее от болезни. Мужчины, унаследовавшие мутацию, заболевают, потому что у них нет второго гена дистрофина, который мог бы восполнить дефектный.
Обычно девочки не испытывают в полной мере последствий МДД, как мальчики, хотя симптомы мышечной слабости у них все же сохраняются. Меньшая часть женщин с мутацией, называемых явными носителями, имеет некоторые признаки и симптомы МДД.
У этих женщин дефицит дистрофина может привести к ослаблению мышц спины, ног и рук, которые быстро утомляются. У явных носителей могут быть проблемы с сердцем, которые могут проявляться в виде одышки или неспособности выполнять умеренные упражнения. Проблемы с сердцем, если их не лечить, могут быть довольно серьезными и даже опасными для жизни.
В очень редких случаях у девочки может полностью отсутствовать вторая Х-хромосома, или ее вторая Х-хромосома могла быть серьезно повреждена. В этих случаях у нее вырабатывается мало или совсем отсутствует дистрофин (в зависимости от типа мутации дистрофина), и у нее развивается дистрофинопатия, как у мальчика.
Родственница мальчика с МДД может пройти полный спектр диагностических тестов, чтобы определить ее статус носительства. Если будет установлено, что она является носителем МДД, регулярные обследования силы и тщательный мониторинг сердца могут помочь ей справиться с любыми симптомами, которые могут возникнуть.
Боль и чувствительность
Уменьшение мышечной массы при МДД само по себе обычно не вызывает болезненных ощущений. Некоторые люди иногда сообщают о мышечных судорогах.
Поскольку мышечная дистрофия не влияет на нервы напрямую, чувствительная сфера не страдает. Также не страдает контроль над гладкими или непроизвольными мышцами мочевого пузыря и кишечника, а также сексуальные функции.
Сердце
Недостаток дистрофина может ослабить сердечную мышцу (миокард), что приведет к состоянию, называемому кардиомиопатией. МДД также может вызывать нарушения проводимости в сердце. Со временем, иногда уже в подростковом возрасте, повреждение сердца, нанесенное МДД, может стать опасным для жизни. Следует внимательно следить за сердцем, обычно у детского кардиолога.
Респираторная функция
Последовательный контроль дыхательной функции следует начинать в возрасте 5-6 лет. Диафрагма и другие мышцы, управляющие легкими, могут ослабнуть, что сделает легкие менее эффективными. Хотя ребенок может не жаловаться на одышку, проблемы, указывающие на плохую дыхательную функцию, включают головные боли, трудности с концентрацией внимания или бодрствования, а также кошмары. Дети, прикованные к инвалидной коляске, обычно имеют признаки плохой легочной функции.
Ослабленные дыхательные мышцы затрудняют кашель, что увеличивает риск серьезной респираторной инфекции. Простая простуда может быстро перейти в пневмонию.
Обучаемость
Около трети мальчиков с МДД имеют некоторую степень нарушения обучаемости, хотя немногие имеют серьезные когнитивные нарушения. Врачи считают, что дистрофиновые нарушения в головном мозге могут незначительно влиять на познание и поведение. Проблемы с обучением при МДД возникают в трех основных областях: концентрация внимания, вербальное обучение и память и эмоциональное взаимодействие.
Диагностика
При диагностике любой формы мышечной дистрофии врач обычно начинает с изучения истории болезни пациента и его семьи и проведения физического обследования. Врачи могут обнаружить псевдогипертрофию, усиление поясничного лордоза, нарушение походки и снижения мышечных рефлексов.
Анамнез пациента и его физическое состояние имеют большое значение для постановки диагноза, даже до того, как будут выполнены какие-либо сложные диагностические тесты.
Кардиомиопатия у пациентов с МДД также может быть связана с нарушениями проводимости. Врач может заметить характерные изменения на электрокардиограмме. Кроме того, с помощью эхокардиографии можно обнаружить структурные изменения в сердце, такие как порок клапанов сердца (особенно поражающий митральный клапан, когда он возникает). Поэтому необходимы электрокардиограмма, неинвазивная визуализация с эхокардиографией или МРТ сердца, а также консультация кардиолога.
Уровни КK и других ферментов
В начале диагностического процесса врачи часто назначают анализ крови, называемый КК (КФК) – креатин(фосфо)киназа, фермент, который выделяется из поврежденных мышц. Когда в образце крови обнаруживается повышенный уровень КФК, это обычно означает, что мышца разрушается в результате какого-либо аномального процесса, такого как мышечная дистрофия или воспаление. Очень высокий уровень КK предполагает, что сами мышцы (а не нервы, которые их контролируют) являются вероятной причиной слабости, хотя это не указывает на то, какой именно тип мышечного расстройства может иметь место. Высокий уровень КФК может быть обнаружен до появления симптомов даже у новорожденных, страдающих МДД.
Уровень КК достигает пика (в 10-20 раз выше верхнего предельного значения) к 2 годам, затем постепенно падает и в конечном итоге возвращается к нормальному уровню, когда значительное количество мышечной ткани заменяется жиром и фиброзной тканью.
Генетическое тестирование
Генетическое тестирование включает анализ ДНК любых клеток (обычно используются клетки крови), чтобы увидеть, есть ли мутация в гене дистрофина, и если да, то где именно она возникает.
Обычно генетическая диагностика показана пациентам с повышенными уровнями КФК в сыворотке и клиническими проявлениями дистрофинопатии. Диагноз подтверждается, если выявлена мутация гена DMD. Генетический анализ в первую очередь направлен на обнаружение крупных делеционных/дупликационных мутаций (в 70–80% случаев присутствуют такие мутации). Если первоначальный генетический анализ отрицательный, следующим следует анализ небольших генных мутаций и генных микроделеций/дупликаций.
Родственницы мужчин и мальчиков с МДД могут пройти ДНК-тестирование, чтобы определить, являются ли они носителями болезни. Женщины, являющиеся носителями МДД, могут передать болезнь своим сыновьям, а их статус носителя — своим дочерям. В меньшинстве случаев девочки и женщины, являющиеся носителями МДД, могут сами проявлять симптомы МДД, такие как мышечная слабость и проблемы с сердцем. Эти симптомы могут не появиться до взрослого возраста.
Несколько экспериментальных препаратов, которые в настоящее время разрабатываются для лечения МДД, требуют знания точной генетической мутации человека, поэтому генетическое тестирование стало важным не только для диагностики, но, возможно, и для будущего лечения.
Биопсия мышц
Для получения более подробной информации, врач может назначить мышечную биопсию, хирургическое удаление небольшой выборки мышц от пациента. Изучая этот образец, врачи могут многое рассказать о том, что на самом деле происходит внутри мышц. Однако в современную эпоху биопсия мышц требуется редко, потому что почти всем пациентам ставится диагноз генетического тестирования.
Терапия
Лекарства, уменьшающие нагрузку на сердце, иногда назначают при МДД.
Лекарства, принадлежащие к группе, известной как кортикостероиды, являются основой фармакологического лечения, поскольку они оказались эффективными в замедлении течения МДД. Детям следует начинать прием этих лекарств до того, как начнется их физическая нагрузка.
Кортикостероиды преднизолон и дефлазакорт полезны при лечении МДД. FDA 9 февраля 2017 г. одобрило дефлазакорт (торговая марка Emflaza), производное оксазолина преднизолона, для лечения МДД. Активность 1 мг преднизолона примерно эквивалентна 1,3 мг дефлазакорта.
Несколько исследований всех этих препаратов при МДД показали значительное увеличение силы (11% при приеме преднизолона по сравнению с плацебо). Это увеличение силы достигло максимума после трех месяцев лечения и сохранялось в течение 18 месяцев; Кроме того, были доказательства улучшения функции мышц (например, время, необходимое для подъема на 4 ступеньки, на 43% быстрее с преднизолоном по сравнению с плацебо) и легочной функции. Кортикостероиды также снижают риск сколиоза и задерживают потерю способности передвигаться. Три исследования показали, что лечение глюкокортикостероидами было связано с улучшением выживаемости. Однако четвертое исследование не показало четкой связи с увеличением выживаемости.
Также было показано, что легочная функция улучшается при лечении преднизолоном по сравнению с плацебо. Форсированная жизненная емкость легких значительно улучшилась (11%) после шести месяцев ежедневного лечения преднизолоном.
Глюкокортикоиды могут задерживать развитие сколиоза и уменьшать необходимость хирургического вмешательства для коррекции сколиоза у пациентов с МДД. Риск развития сколиоза может быть значительно ниже у пациентов, получающих ежедневное лечение дефлазакортом, по сравнению с плацебо, а необходимость в хирургии позвоночника также значительно снижается у пациентов, принимающих дефлазакорт. Распространенность переломов у пациентов, получающих глюкокортикоиды, и тех, кто их не лечит, одинакова. Есть некоторые свидетельства того, что лечение глюкокортикоидами при МДД улучшает выживаемость, однако другие данные не показывают связи между выживаемостью и лечением глюкокортикоидами.
Постоянное применение кортикостероидов является частью стандартной медицинской помощи при МДД, но такое лечение может привести к побочным эффектам, таким как увеличение веса, низкий рост, угри, изменения поведения, остеопороз, компрессионные переломы длинных костей и позвонков. Уместно контролировать пациентов, получающих кортикостероидную терапию, с периодической визуализацией позвоночника, поскольку они могут протекать бессимптомно. Детей, у которых развиваются переломы позвонков или длинных костей, следует направлять к детскому эндокринологу или специалисту по костям.
Быстрая отмена кортикостероидов может привести к опасным для жизни осложнениям.
В сентябре 2016 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) предоставило ускоренное одобрение препарата Этеплирсен, препарата для пропуска экзона, который, как было показано, повышает уровень дистрофина у пациентов с мутацией гена дистрофина, поддающейся пропуску экзона 51.
Аталурен (также известный как PTC124) — это пероральный препарат, разрабатываемый для лечения генетических дефектов, вызванных бессмысленными мутациями, позволяющий обойти бессмысленную мутацию и продолжить процесс трансляции до производства функционирующего белка, что было продемонстрировано в нескольких исследованиях. исследования. Такой подход может принести пользу примерно от 10% до 15% пациентов с МДД / МПК, которые имеют бессмысленные (стоп-мутации) мутации. Аталурен лицензирован в Европейском Союзе и Великобритании для лечения пациентов в возрасте 2 лет и старше с МДД, вызванным бессмысленными мутациями.
Наиболее частым побочным эффектом аталурена является рвота. Другие включают снижение аппетита, потерю веса, головную боль, гипертонию, кашель, кровотечение из носа, тошноту, боль в верхней части живота, метеоризм, дискомфорт в животе, запор, сыпь, боль в конечностях, скелетно-мышечную боль в груди, кровь в моче, непроизвольное мочеиспускание и лихорадку.
В декабре 2019 года Vyondys 53, препарат для «пропуска экзона», нацеленный на участок ДНК, называемый экзоном 53, был одобрен FDA для лечения лиц с подтвержденной мутацией гена DMD, поддающейся терапевтической стратегии под названием экзон 53. пропускать и может помочь до 8% людей с МДД.
В августе 2020 года Viltepso, препарат для «пропуска экзона», нацеленный на участок ДНК, называемый экзоном 53, был одобрен FDA для лечения людей с подтвержденной мутацией гена DMD, поддающейся терапевтической стратегии под названием экзон 53. пропускать и может помочь до 8% людей с МДД.