Кінцівко-поясна м’язова дистрофія (КПМД)
Кінцівко-поясна м’язова дистрофія (КПМД) – це різноманітна група захворювань з безліччю підтипів, які класифікуються за геном захворювання і спадковості. КПМД зазвичай проявляється слабкістю в проксимальних м’язах стегон та плечей, виникає після досягнення здатності самостійно пересуватися.
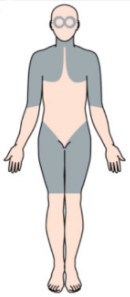
Плечовий пояс – сукупність кісток (пари лопаток і ключиць) і м’язів, що забезпечують опору і рух верхніх кінцівок, а тазовий пояс – складається з кісток стегна (клубової кістки, сідничної кістки і лобка), згрупованих в кільце, і з’єднує область таза хребта з нижніми кінцівками. В сукупності вони називаються поясами кінцівок, і саме в них спостерігається слабкість і атрофія (виснаження) м’язів, пов’язаних з поясами кінцівок, дали назву цій групі захворювань. Проксимальні м’язи – це ті, які знаходяться найближче до центру тіла; дистальні м’язи знаходяться далі від центру.
Поширеність КПМД
КПМД є четвертою за поширеністю генетичної причиною м’язової слабкості, з передбачуваною поширеністю всіх підтипів КПМД від 4 до 7 на 100 000, в залежності від географічного та етнічного походження.
Успадкування КПМД
Це група генетичних захворювань. КПМД може бути успадкована одним з двох основних способів, які відомі як аутосомно-домінантний і аутосомно-рецесивний тип спадкування. Слово «аутосомний» означає, що задіяні гени не перебувають на хромосомі X або Y і, отже, передаються від чоловіків, жінок або від обох.
При захворюваннях з домінантним типом успадкування людина, що успадкувала дефектний ген від одного з батьків, матиме симптоми хвороби. У цього батька теж була хвороба. При захворюваннях з рецесивним успадкуванням людина повинна успадкувати два дефектних гена – по одному від кожного з батьків, щоб мати симптоми хвороби. У батьків немає симптомів.
Рецесивна форма КПМД може проявитися у однієї людини, коли немає сімейного анамнезу. Інші члени сім’ї могли бути носіями без симптомів хвороби. Носії мають генетичний дефект (мутацію) на хромосомі і можуть мати дитину з цим захворюванням, але тільки якщо другий з батьків дитини також є носієм. Таким чином, носії рідкісного рецесивного захворювання нерідко не знають, що вони є носіями, до тих пір, поки у когось з членів сім’ї не розвинеться хвороба.
У людини з КПМД може бути абсолютно нова генетична мутація, тому в сім’ї дійсно може не бути сімейного анамнезу або навіть носіїв розладів. Однак, як тільки у когось розвивається генетичне захворювання, навіть якщо мутація є спонтанною (нової) у цієї людини, він або вона може передати мутацію будь-якому потомству, тим самим впроваджуючи ген захворювання в сім’ю.
Причина КПМД
Гени, пов’язані з КПМД, зазвичай кодують білки, які відіграють життєво важливу роль в м’язовій функції, регуляції та відновленні. Коли один з цих генів містить мутацію, клітини не можуть виробляти білки, необхідні для здорових м’язів. Існує близько 35 форм кінцівко-поясної м’язової дистрофії (КПМД) і класифікуються вони за генетичними дефектами, які їх викликають.
Гени, розташовані на хромосомах в кожній клітині тіла, представляють собою коди або рецепти для виробництва різних білків організму. Гени, пов’язані з КПМД, зазвичай виробляють білки, необхідні для роботи м’язів. Коли проблеми з білком виникають через несправність одного з цих генів, волокна в м’язах не працюють належним чином. Поступово м’язи стають настільки слабкими, що люди відчувають симптоми КПМД. Оскільки КПМД прогресує, м’язи продовжують слабшати протягом усього життя людини.
Деякі з генів, які, будучи пошкодженими, викликають КПМД, призводять до вироблення білків, які зазвичай розташовані в мембрані м’язових клітин, тонкій оболонці, яка оточує кожну м’язову клітку, допомагаючи захистити її від травм під час скорочення м’язів. Якщо який-небудь з цих білків відсутня, мембрана втрачає деякі зі своїх «амортизуючих» властивостей, і їй стає важче захищати м’язову клітку від пошкоджень під час нормальних циклів скорочення і розслаблення.
Виділяють дві основні групи КПМД, іменовані КПМД1 і КПМД2, за відповідними типами успадкування: аутосомно-домінантний (КПМД1) і аутосомно-рецесивний (КПМД2).
Мутації в десятках різних генів викликають певні підтипи КПМД1 і КПМД2. У цих випадках білки, пов’язані з цими генами, нефункціональні або недостатньо функціональні, і м’язи не можуть нормально функціонувати. Поступово м’язи стають настільки слабкими, що люди відчувають симптоми КПМД.
На додаток до відомих підтипів КПМД1 і КПМД2, пов’язаних з конкретними генами, існує багато випадків КПМД, для яких ген, що викликає захворювання, ще не відомий (і люди з цими випадками не ідентифіковані як такі, що мають підтип-специфічну форму КПМД). Вчені активно працюють, щоб зрозуміти причини цих неідентифікованих підтипів, тому що чим більше ми розуміємо різні причини КПМД і різні способи, якими можуть бути скомпрометовані м’язи, тим більше шансів знайти ефективні методи лікування, щоб втрутитися в патологічний процес.
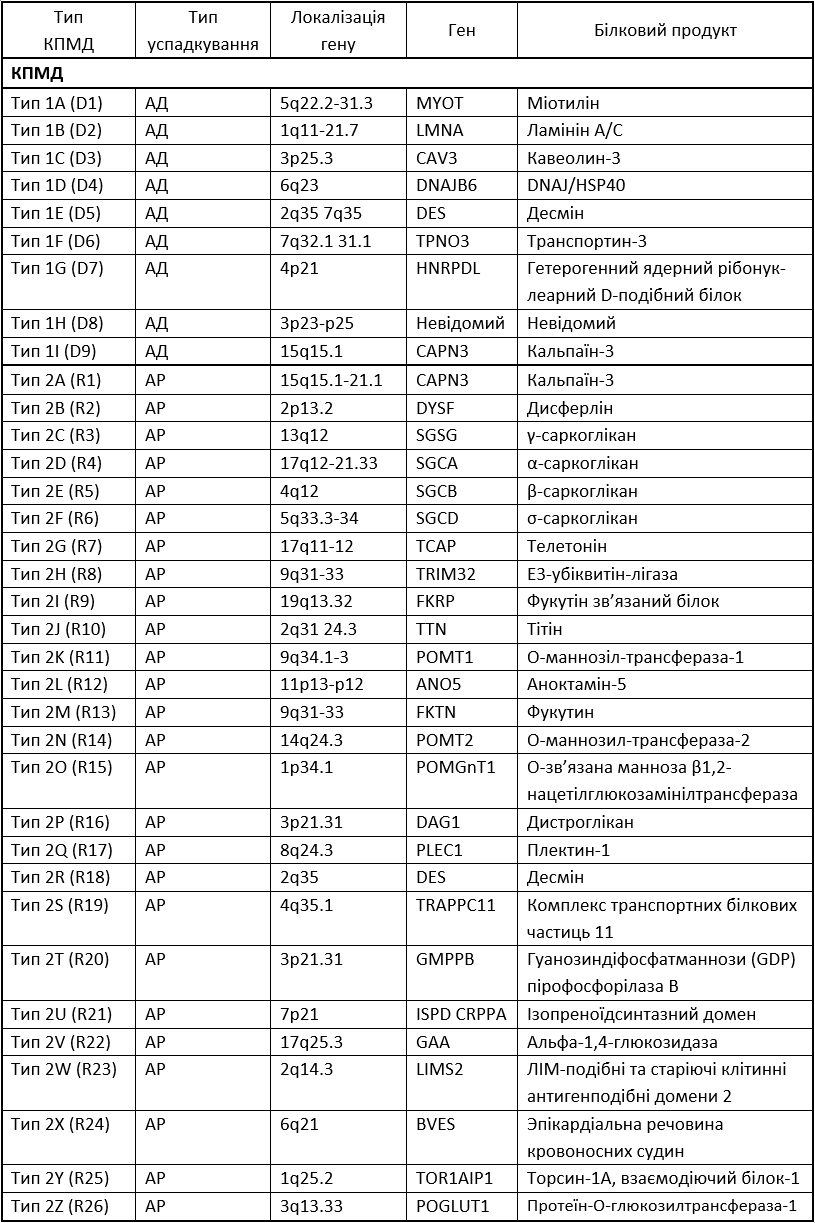
Підтипи КПМД
Нижче наведено список підтипів КПМД, відомих на даний час із зазначенням типу успадкування, гена і його білкового продукту, локалізації гена.
Симптоми КПМД
Основними клінічними та об’єднуючими ознаками КПМД є прогресуюча слабкість та атрофія м’язів, що в основному вражає плечовий пояс (плечовий тип), тазовий пояс (тазофеморальний тип) або і те, і інше. Більшість випадків захворювання, що почалося в дитинстві, мають тазофеморальну слабкість. Захворювання, що почалося у дорослих, зазвичай вражає як плечовий, так і тазовий пояс з поступово наростаючою слабкістю проксимальних відділів кінцівок. Слабкість м’язів обличчя зазвичай буває легкої або повністю відсутній.
Екстраокулярні м’язи (м’язи, які контролюють рух очі) не пошкоджуються при КПМД. Сила дистальних м’язів зазвичай зберігається навіть на пізній стадії захворювання, але слабкість дистальних м’язів може бути різною або ведучою ознакою деяких підтипів КПМД.
КПМД2 являє собою велику клінічну гетерогенність, більш пізній вік початку, більш поступове прогресування і підвищення креатинкінази, яке може бути мінімальним у порівнянні з КПМД1.
Часто люди з КПМД вперше помічають проблему, коли починають ходити «перевальцем», так звана «качина» хода, через слабкість м’язів стегон та гомілок. Їм може бути важко вставати зі стільця, вставати з сидіння унітазу або підніматися по сходах. По мірі прогресування слабкості людині може знадобитися використання допоміжних засобів пересування.
Слабкість в плечовій області може ускладнювати утримання витягнутих рук або пересування важких предметів. Може стати все важче утримувати руки над головою під час розчісування волосся або розстановці речей на високій полиці. Деяким людям важче набирати текст на комп’ютері, і у них навіть можуть виникнути проблеми з харчуванням.
Допоміжні пристосування, такі як тростина, можуть полегшити завдання по мірі прогресування слабкості. Коляска або самокат з електроприводом стають зручними, коли слабкість в тазовому поясі і верхній частині ніг викликає часті падіння.
Деякі з підтипів КПМД також характеризуються додатковими симптомами. Наприклад, при деяких типах КПМД може вражатися серце, але це не відбувається так часто, як це відбувається при деяких інших формах м’язової дистрофії. Проблеми з серцем можуть приймати дві форми – слабкість серцевого м’яза (кардіоміопатія) і порушення передачі сигналів, що регулюють серцебиття (порушення провідності або аритмії). Необхідно контролювати серце на предмет цих ускладнень. При необхідності для їх лікування можна використовувати ліки або навіть спеціальні пристрої (наприклад, кардіостимулятори).
Деякі підтипи КПМД також пов’язані з м’язами, використовуваними для дихання, і з цієї причини слід регулярно контролювати дихальну (респіраторну) функцію, яка згодом може знижуватися.
Інші симптоми можуть бути присутніми при деяких різних підтипах КПМД, включаючи, крім іншого, контрактури суглобів, м’язові спазми, збільшення в обсязі литкових м’язів і залучення дистальних м’язів рук і ніг.
КПМД, як і інші м’язові дистрофії, в першу чергу є захворювання довільних м’язів. Це м’язи, які люди використовують для руху кінцівок, шиї, тулуба та інших частин тіла, які перебувають під довільним контролем. Мимовільні м’язи, за винятком серця (який є особливим типом мимовільних м’язів), при КПМД не зачіпаються. Травлення, функції кишечника і сечового міхура і сексуальна функція, які виконуються мимовільними м’язами, залишаються в нормі.
Біль не є характерним симптомом при КПМД, хоча обмежена рухливість іноді призводить до хворобливості м’язів і болів у суглобах. Вправи, що дозволяють зберігати гнучкість суглобів, як можна більше рухатися, теплі ванни і, при необхідності, ліки можуть звести цей дискомфорт до мінімуму.
Мозок, інтелект і чутливість зазвичай не страждають при КПМД. Хоча при деяких підтипів КПМД, спостерігається розумова відсталість.
Прогресування КПМД
В даний час неможливо з впевненістю передбачити прогресування кожного типу КПМД, хоча може виявитися корисним знання того, що лежить в основі генетичної мутації. Деякі форми розладу прогресують до втрати здатності ходити протягом декількох років і викликають серйозну інвалідність, в той час як інші прогресують дуже повільно протягом багатьох років і викликають мінімальну інвалідність.
КПМД може початися в дитинстві, підлітковому віці, юності або навіть пізніше. Обидві статі страждають однаково.
Є думка, що, якщо КПМД починається в дитинстві, прогресування зазвичай відбувається швидше, а хвороба стає більш тяжкої. Якщо розлад починається в підлітковому або в дорослому віці, як правило, воно не таке серйозне і прогресує повільніше. Перебіг зазвичай являє собою повільно прогресуючу, в основному симетричну слабкість, за винятком кількох типів з швидким прогресуванням або асиметричною слабкістю.
Діагностика
При діагностиці будь-якої форми м’язової дистрофії лікар зазвичай починає з вивчення історії хвороби пацієнта і його сім’ї і проведення фізичного обстеження. Анамнез і фізичний стан яких багато важать для постановки діагнозу, навіть до того, як будуть проведені будь-які лабораторні тести. (див. Діагностика).
Лабораторні дослідження
Концентрація креатинкінази (КК) у сироватці зазвичай помірно підвищена при КПМД. Однак він може бути дуже високим при деяких підтипів КПМД, таких як саркогліканопатія, дісферлінопатія і кавеолінопатія.
Медична допомога
Тактика при м’язовій дистрофії кінцівок (КПМД) є підтримуючої; терапія, модифікуюча перебіг хвороба, відсутня. Цілі терапії включають підтримку рухливості і функціональної незалежності, усунення пов’язаних з цим ускладнень і максимальне поліпшення якості життя. Для забезпечення оптимального догляду рекомендується мультидисциплінарне лікування в центрах, що мають досвід лікування нервово-м’язових захворювань.
Допоміжні пристрої
Прості пристосування, такі як тростину, можуть полегшити ходьбу і виконання завдань у міру прогресування слабкості. Коляска або самокат з електроприводом стають зручними, коли слабкість в тазовому поясі і стегні викликає падіння. Люди, у яких КПМД досягла цієї стадії, часто виявляють, що велика частина їх незалежності повертається, і стомлюваність значно знижується, коли вони починають використовувати цей тип мобільного обладнання.
Серце, дихання і харчування
Серце може бути порушено при деяких підтипів КПМД, зокрема при КПМД1B, КПМД2A-I, але це не відбувається так часто, як це відбувається при інших формах м’язової дистрофії. Серце слід контролювати на предмет ускладнень за допомогою огляду, електрокардіограми (ЕКГ) і структурних досліджень серця з допомогою ехокардіографії (ЕхоКГ) або магнітно-резонансної томографії серця (МРТ). При необхідності для лікування серцевих ускладнень можна використовувати ліки або навіть пристрої (наприклад, кардіостимулятори). Трансплантація серця може знадобитися тим, у кого розвинулася важка застійна серцева недостатність.
Дихальна функція може з часом знижуватися, і це теж слід регулярно контролювати. Деякі КПМД (наприклад, КПМД2I) пов’язані зі слабкістю дихальних або ротоглоткових м’язів і підвищеним ризиком дихальної недостатності з прогресуванням захворювання. Пацієнтам з КПМД, у яких є ознаки дихальної недостатності, денної сонливості або симптоми порушення дихання уві сні, може бути корисна неінвазивна вентиляція, яка включає пристрої, які можуть допомогти підтримувати дихальну функцію.
Дисфагія (утруднення ковтання) і слабкість рук, пов’язані з КПМД, можуть привести до недоїдання. Пацієнти з недостатнім споживанням їжі, проблемами з ковтанням, аспірацією (коли їжа, слина, рідини або блювота вдихаются в легені, а не проковтують в стравохід) або з втратою ваги повинні бути оцінені за допомогою досліджень ковтання або спрямовані до гастроентеролога. У таких пацієнтів можуть бути корисні методи, що поліпшують ковтання, такі як зміна консистенції їжі (використання загусників їжі), використання маневру підборіддя або установка зонда для годування.
Не існує ніяких спеціальних дієтичних обмежень або добавок, які б безпосередньо впливали на перебіг КПМД. Лікар може порекомендувати деяким людям дієту для зниження або стабілізації ваги, оскільки значний надмірна вага створює велике навантаження на і без того ослаблені м’язи.
Терапія і вправи
Програми фізіотерапії та трудотерапії зазвичай є частиною лікування КПМД. Трудотерапія фокусується на певних діях і функціях, особливо на використанні рук, в той час як фізіотерапія підкреслює рухливість і (де це можливо) зміцнення великих груп м’язів. Лікар нашого центру може направити на консультацію до ерготерапевта для ретельної оцінки та індивідуальної програми вправ.
Основні цілі фізіотерапії – забезпечити більшу рухливість суглобів і запобігти контрактури. Ці проблеми можуть виникати, коли рух обмежено і для комфорту пацієнта і його функцій важливо уникати їх. Тому важливо, щоб пацієнти продовжували рухатися якомога більше.
У працетерапії основна увага приділяється поліпшенню здібностей, пов’язаних з роботою, відпочинком або повсякденним життям. Наприклад, опори для рук можуть робити такі завдання, як використання комп’ютера або укладання волосся, менш нудними.
При КПМД певні види вправ, що викликають стрес, можуть прискорити пошкодження м’язів. Пацієнтам з м’язовою дистрофією слід підтримувати адекватну гідратацію; уникайте понад максимальних вправ високої інтенсивності; і уникайте тренувань до знемоги.
Деякі фахівці рекомендують плавання і водні вправи як хороший спосіб підтримувати м’язи в тонусі, наскільки це можливо, не викликаючи надмірного стресу. Плавучість води допомагає захистити від певних видів м’язової напруги і травм. Перед тим, як приступити до програми вправ, переконайтеся, що ви пройшли оцінку серця, і не плавайте поодинці.
Ортопедичні ускладнення
Пацієнти з КПМД мають підвищений ризик розвитку деформацій хребта, в тому числі кіфозу (горбатий типу викривлення) або сколіозу (бічне відхилення хребта). Пацієнтів, у яких розвиваються деформації хребта, слід направляти до фахівця ортопеда-травматолога / нейрохірурга для обстеження і операції, якщо необхідно, для підтримки оптимальної постави, рухливості, серцевої і дихальної функції.